
STSCI 4060 Final Project
代写Python作业 In this project you will have an opportunity to integrate Python programming, Oracle database, database-driven dynamic web pages
(Due: May 18, 2022)
Important: Read and follow this whole document carefully!
How to submit: submit your project report to the Canvas course website with a single zip file, which combines all your files.
General instructions: 代写Python作业
• Do your own work. Any cheating behavior (for example, submitting code similar to that of other student(s), copying code from an Internet source, etc.) may result in a serious consequence (e.g., getting zero points, failing the class, …). If you have a question about the project, you should directly email your instructor.
• Start the project early. Programming is time consuming; you will need significant amount of time and patience to code some portions of the project. Do not expect to finish it on the due day. 代写Python作业
• Test your code (especially the .cgi files) separately from other systems. When you have multiple software systems connected together, it is hard to debug.
• Add sufficient documentation to your code so that people understand your algorithm and what your code does. This is a requirement of this project.
• Do not edit the raw data file in any way. Your results will be compared to the standard solutions.
• Make sure that you have included all the components in your submission (see the details at the end of this document on pages 3 and 4). Your grader will run your programs on his/her computer; if something is missing your programs will not run. 代写Python作业
In this project you will have an opportunity to integrate Python programming, Oracle database, database-driven dynamic web pages, and Python data analysis modules with Jupyter (IPython) notebook using the data that are processed with the above integration. You are given a raw data file,honeybee_gene_sequences.txt, which was downloaded from the NCBI web site. We dealt with the protein data in the class; however, genes are different kinds of biomolecules. Unlike proteins that are composed of 20 amino acids, genes are only formed with four building elements: adenine (A), cytosine (C), guanine (G) and thymine (T).
They are called nucleotides, a sequence of which forms a gene, which then determines the sequence of a protein. Thus, the compositions of the nucleotides and their relative frequencies, especially the combined relative frequency of C and G (i.e., the sum of the percentages of C and G in a gene sequence), have important biological (or medical) meanings. For this project, you will do the following: 代写Python作业
- Design a web page (using KompoZer or another program) to allow a user to enter a file name (here honeybee_gene_sequences.txt) and the full path to the location where the file is stored so that the user can upload the data file by clicking the Submit button on the web page.
Write a specific .cgi file with Python to accept the user input from the web page, process the data and store the processed data in an Oracle database table, which is also created within the .cgi file using the Python-Oracle integration approach. In this .cgi file, you need to at least include the following functions:
A. The main() function to receive the user input from the web page.
B. The processInput() function to do the following:
a) Read in the contents of the data file. 代写Python作业
b) In order to extract the right nucleotide (or gene) sequences for all possible cases (you can see that most times the nucleotide sequences start right after the substring, mRNA, but not always), you are required to insert the substring, _**gene_seq_starts_here**_, right before the nucleotide sequences of every bee gene (or entry) through Python programming when you read in (or process) the raw data line by line. In this way, you will use the _**gene_seq_starts_here**_ substring as the starting point to extract the nucleotide sequences later on. 代写Python作业
c) Extract the gi number and nucleotide sequence of each gene (or entry).
d) Make sure that your Python program correctly reads in the gene (or nucleotide) sequence of the last entry in the raw data file.
e) Calculate the relative frequencies of each nucleotide in every gene.
f) Calculate the combined relative frequency of the nucleotides G and C, freq_GC, which is obtained by adding the relative frequencies of G and C.
g) Connect Python to the Oracle database system. 代写Python作业
h) Create an Oracle table called beeGenes to store gi numbers, nucleotide sequences, the relative frequencies of the four nucleotides and the combined relative frequencies of the nucleotides G and C, freq_GC. So, your beeGenes table has seven columns.
i) When you write the data to the database table, you are required to use the Oracle bind variable approach and the batch writing method by setting the bindarraysize to a certain number (refer to the lecture slides if needed).
j) In order not to truncate any gene sequence, you need to find an appropriate number for the sequence input size. Thus, you are required to write a separate Python program (which should also be submitted for grading) to determine the maximum number of nucleotides of all the genes in the data file.
C. fileToStr() to return a string containing the contents of the named html file.
D. makePage() to make the final formatted string (or webpage) for displaying on a web page. 代写Python作业
- Design a template web page to acknowledge that the uploading process was successful and that the data were processed and stored in the database as planned. There is a button on which a user can click if the user wants to see some results, retrieved from the Oracle database table you just created.
Code another .cgi file with Python to retrieve data from the database table (beeGenes). The functions you need are similar to those in the previous .cgi file, but in the processInput() function you are required to use a Python dictionary and the format string mechanism when you extract data from beeGenes. In this function, you will run queries against the beeGenes table to find the gi numbers of those bee genes that have the highest relative frequencies of nucleotide A, C, G, or T so that you can display these on the final web page when the user clicks the “Click to See Some Result” button on the confirmation page of data submission.
5. Design another template web page to display the results gathered from the database. Inserting a hyperlink of the nucleotides to another web page is optional.
You use the local server to run all the web services in this project, using port number 8081. 代写Python作业
Write a Python program to run a query against the Oracle table beeGenes to show that you earlier successfully extracted the gene sequence of the last entry of the raw data file. To do so, you run a query for the gene sequence by providing the related gi number, which is 147907436. Include both your Python code and the query result in your report.
Connect Python to the Oracle database and conduct a K- Means cluster analysis in a Jupyter notebook. You should only use three columns in the beeGenes table : freq_A (relative frequency of the nucleotide A), freq_T (relative frequency of the nucleotide T) and freq_GC for this analysis due to some biological reasons.
In your Jupyter notebook, you should use three cells: the 1st cell is for importing all the necessary Python modules for this analysis; the 2nd cell is to connect Python to your Oracle database and create a numpy array containing the three columns of data that are read from the beeGenes table in your Oracle database; and the 3rd cell is for carrying out the K-Means analysis and plotting a 3D scatter plot using the three columns of data based on the clusters identified by the K-Means analysis. 代写Python作业
The K-Means settings are: n_cluster=7, init='random', n_init=10, max_iter=500, tol=1e-4, and random_state=0. Then, you create a scatter plots with a total figure size of 14X14. Use the same type of marker ('o') for all the clusters, set s to 20, set labels to "Cluster 1" to "Cluster 7" for the cluster values of 0 to 6 that are found by the K-Means algorism, respectively. Set the colors as follows: red for Cluster 1, blue for Cluster 2, aqua for Cluster 3, black for Cluster 4, purple for Cluster 5, magenta for Cluster 6, and green for Cluster 7. 代写Python作业
Mark the centroid of each cluster with a star : set s to 100, color to red and label to Centroids. Give the title "K-Means" to the plot. The legends should be displayed in the upper right corner of the plot.
After your code works correctly, run all the cells in your Jupyter notebook at once. Submit the notebook file (.ipynb) and an HTML file of the same notebook (.html).
Your report should at least contain the following items: all your code, outputs and screenshots. Which must be combined into a single PDF file, arranged in the order they appear in the project. You must mark all your items clearly. Moreover, your Python and html program files must be submitted as separate files, which must be in the same folder (no subfolders) so that your grader can run your programs successfully. The following is a detailed list of the files/items to submit. 代写Python作业
• All Python program files (with the .py extension), including the program to find the maximum number of nucleotides in a gene sequence and the program to query the database to confirm that you successfully extracted the gene sequence of the last entry of the raw data file.
• All .cgi files, which are technically Python files but contain the .cgi extension.
• All .html files, including the template and non-template .html files.
• The design window of your input web page.
• The design windows of your two template web pages.
• A screenshot of your input web page with the input value entered 代写Python作业.
• A screenshot of your confirmation web page that displays that you have successfully submitted the data, etc.
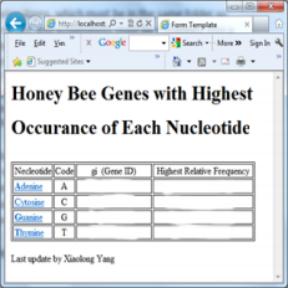
• A screenshot of your final web page that displays the results of database query similar to the following screenshot (but it is only an example, here, and the actual results were erased).
• A screenshot of the local CGI server log.
• The result of Oracle table query for the gene sequence of the last entry, which should be a Python shell screenshot (you may need more than one screen to display the complete sequence).
• Your Jupyter notebook file (.ipynb).
• The Jupyter notebook HTML file (.html). 代写Python作业
• The localCGIServer.py file.
• The raw data file, honeybee_gene_sequences.txt.
************************** Have fun doing this project! ************************

更多代写:cs代写 经济代考 会计代写 计算机科学代写 心理学essay代做


发表回复
要发表评论,您必须先登录。